用R和BioConductor进行基因芯片数据分析(五):芯片间归一化
May 05, 2008上次进行了芯片内的归一化,但是我们的数据来自于10张芯片,为了让这10张芯片之间有可比性,需要进行芯片间归一化。
由于我比较懒,具体原理就不介绍了。
这里用到Bioconductor的一个package,叫做limma,以及其中的函数normalizeBetweenArrays()
由于normalizeBetweenArrays()需要log intensity或log ratio作为输入,于是先进行log转化:
#log transformation norm_log<-matrix(data = NA, nrow =dim(normed)[1], ncol = dim(normed)[2], byrow = TRUE, dimnames = NULL) for (i in 1:dim(normed)[1]){ for (j in 1:dim(normed)[2]){ norm_log[i,j]<-log(normed[i,j])/log(2) } }然后利用函数进行芯片间归一化:
library(limma) norm_log_btw_array<-normalizeBetweenArrays(norm_log,method='scale')normalizeBetweenArrays()函数有许多方法,具体请看帮助。
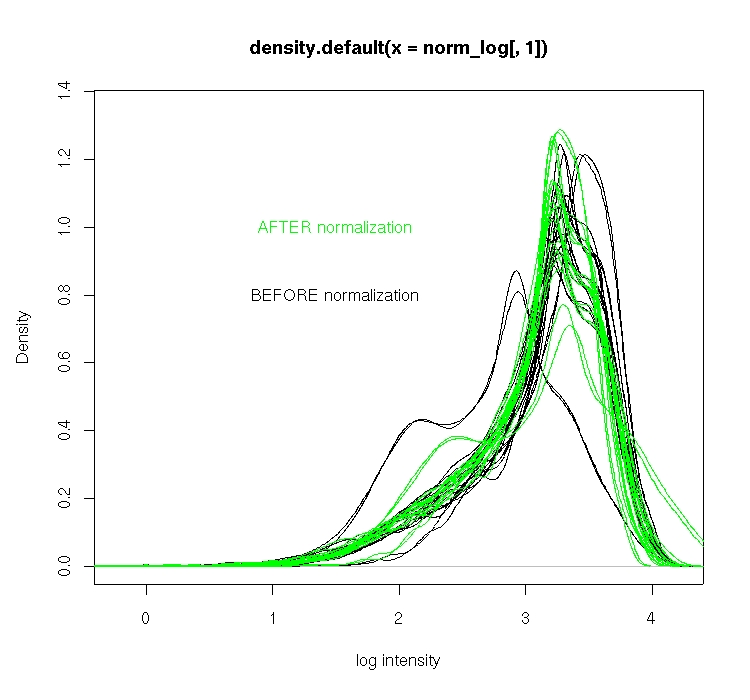
下面看看效果吧
plot(density(norm_log[,1]),ylim=c(0,1.35),xlab='log intensity') for (i in 2:20){ lines(density(norm_log[,i]),type='l') } lines(density(norm_log_btw_array[,1]),type='l',col='green') for (i in 2:20){ lines(density(norm_log_btw_array[,i]),type='l',col='green') } text(1.5,c(0.8,1.0),labels=c('BEFORE normalization','AFTER normalization'),col=c('black','green'))